- CATALYSE - Catalyse homogène
- CATALYSE - Catalyse homogèneLa plupart des réactions chimiques requièrent, pour avoir lieu, un apport d’énergie, l’énergie d’activation. Certaines substances sont susceptibles de diminuer cette énergie d’activation et favorisent donc la réaction; elles interviennent par une ou plusieurs des fonctions suivantes: disposition des partenaires réactionnels dans un arrangement énergétiquement et stériquement favorable, levée des restrictions imposées par les règles de sélection des orbitales moléculaires, introduction d’un nouveau chemin réactionnel efficace grâce à des interactions spécifiques entre les partenaires réactionnels et elles-mêmes.Ces substances sont appelées catalyseurs lorsqu’elles sont récupérées en fin de réaction, ce qui les distingue des autres partenaires réactionnels que sont les substrats et réactifs. Le phénomène correspondant, la catalyse, a été reconnu au XIXe siècle, mais est resté pendant longtemps très mal compris. Au début du XXe siècle, l’étude des réactions catalytiques se développa de façon pragmatique et très empirique. Ces études furent d’abord centrées sur ce qu’il est convenu d’appeler la catalyse de contact, ou hétérogène, les partenaires réactionnels et le catalyseur solide interagissant au sein d’un milieu hétérogène.La possibilité d’obtenir en solution une action catalytique semblable, par l’intermédiaire de solutés acides ou basiques, mais aussi de certains ions métalliques, est apparue à partir de 1925. L’importance de la catalyse homogène s’est accrue, au cours des dernières décennies, du fait du développement très rapide des connaissances scientifiques dans les domaines des acides, des bases, des composés de coordination et des composés organométalliques, et, d’autre part, des applications de plus en plus nombreuses dans des domaines aussi variés que la pétrochimie, la polymérisation et la synthèse organique. La première action du catalyseur sur un substrat qui va réagir est souvent une perturbation de ce dernier par le centre catalytique; cette perturbation fait intervenir un échange électronique et correspond donc à une interaction acide-base selon le concept de Lewis:
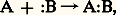 où A représente un acide, c’est-à-dire un composé pouvant accepter une paire d’électrons, et:B, une base, c’est-à-dire un composé susceptible de céder une paire d’électrons. Les actions acide-base peuvent être classées en dures et molles et mettent préférentiellement en jeu des couples acide et base soit durs, soit mous. Selon Pearson, les bases molles sont, qualitativement, celles dans lesquelles l’atome donneur possède une forte polarisabilité, une faible électronégativité et des orbitales externes complètes; de même, les acides mous possèdent une charge positive faible, une grande taille et des orbitales externes vacantes. Les bases dures et les acides durs ont des propriétés opposées.Dans cette perspective, on peut donc distinguer une catalyse dure, qui fait intervenir un centre catalytique polarisant fortement le substrat, et une catalyse molle, qui met en jeu la formation d’une liaison entre le centre catalytique et le substrat ne conduisant pas à une forte séparation des charges. Dans le premier cas, les centres catalytiques sont le proton, les ions des métaux non de transition mais aussi des ions des métaux de transition dans un état d’oxydation élevé; les propriétés catalytiques de ces centres sont liées à leur faible polarisabilité et à leur grand pouvoir polarisant: la catalyse acidobasique entre dans cette catégorie. Dans le second cas, l’échange d’électrons entre le substrat et le centre catalytique prédominera sur les interactions électrostatiques; les centres catalytiques sont constitués par les métaux non de transition réduits et les métaux de transition dans leurs états d’oxydation faibles: la catalyse de coordination, qui utilise des complexes de coordination comme catalyseurs, constitue le domaine le plus important de la catalyse molle. Elle est souvent confondue avec la catalyse homogène, mais ce terme, dans l’optique de la présente introduction, constitue une contraction abusive de «catalyse homogène par les composés de coordination des métaux de transition».Dans certains cas, la catalyse dure ou molle par les métaux, de transition ou non, a pour effet de transférer des électrons entre les partenaires réactionnels. Lorsque le transfert est monoélectronique, elle conduit à la formation de radicaux qui réagissent ultérieurement selon les processus classiques de la chimie radicalaire. On désignera par catalyse redox ce type particulier de catalyse.L’importance de la catalyse homogène dans l’industrie chimique est difficile à déterminer parce que ses domaines d’application sont en constante évolution. Ainsi, la catalyse acido-basique voit son importance diminuer, alors que les catalyseurs redox et de coordination prennent parfois le relais des catalyseurs hétérogènes. L’introduction de catalyseurs immobilisés sur un support contribue aussi à cette difficulté et il serait préférable de considérer dans cet article la catalyse moléculaire par opposition – schématique – à la catalyse hétérogène. La nature moléculaire de ces catalyseurs autorise leur emploi dans des conditions plus douces et favorise des transformations plus efficaces. De ce fait, ils sont générateurs d’économies d’énergie et de matières premières, facteurs dont il faudra de plus en plus tenir compte.1. Catalyse acido-basiqueLes acides et les bases sont les plus simples des catalyseurs de la phase liquide et les plus anciennement connus. Plusieurs définitions leur sont associées. Selon Brønsted, un acide est une substance qui a tendance à perdre un proton; une base est une substance capable de capter un proton. La définition de Lewis englobe les acides de Brønsted ainsi que tous les composés pouvant donner ou accepter des paires d’électrons [cf. ACIDES ET BASES].Dans les réactions de catalyse acido-basique, l’une au moins des étapes est un transfert de proton du catalyseur au substrat S (catalyse acide) ou du substrat au catalyseur (catalyse basique). Le substrat intervient, de ce fait, soit comme base, soit comme acide, et la réaction implique donc un équilibre protolytique. Plusieurs possibilités peuvent apparaître. En catalyse acide:– l’équilibre protolytique est l’étape lente du processus:
où A représente un acide, c’est-à-dire un composé pouvant accepter une paire d’électrons, et:B, une base, c’est-à-dire un composé susceptible de céder une paire d’électrons. Les actions acide-base peuvent être classées en dures et molles et mettent préférentiellement en jeu des couples acide et base soit durs, soit mous. Selon Pearson, les bases molles sont, qualitativement, celles dans lesquelles l’atome donneur possède une forte polarisabilité, une faible électronégativité et des orbitales externes complètes; de même, les acides mous possèdent une charge positive faible, une grande taille et des orbitales externes vacantes. Les bases dures et les acides durs ont des propriétés opposées.Dans cette perspective, on peut donc distinguer une catalyse dure, qui fait intervenir un centre catalytique polarisant fortement le substrat, et une catalyse molle, qui met en jeu la formation d’une liaison entre le centre catalytique et le substrat ne conduisant pas à une forte séparation des charges. Dans le premier cas, les centres catalytiques sont le proton, les ions des métaux non de transition mais aussi des ions des métaux de transition dans un état d’oxydation élevé; les propriétés catalytiques de ces centres sont liées à leur faible polarisabilité et à leur grand pouvoir polarisant: la catalyse acidobasique entre dans cette catégorie. Dans le second cas, l’échange d’électrons entre le substrat et le centre catalytique prédominera sur les interactions électrostatiques; les centres catalytiques sont constitués par les métaux non de transition réduits et les métaux de transition dans leurs états d’oxydation faibles: la catalyse de coordination, qui utilise des complexes de coordination comme catalyseurs, constitue le domaine le plus important de la catalyse molle. Elle est souvent confondue avec la catalyse homogène, mais ce terme, dans l’optique de la présente introduction, constitue une contraction abusive de «catalyse homogène par les composés de coordination des métaux de transition».Dans certains cas, la catalyse dure ou molle par les métaux, de transition ou non, a pour effet de transférer des électrons entre les partenaires réactionnels. Lorsque le transfert est monoélectronique, elle conduit à la formation de radicaux qui réagissent ultérieurement selon les processus classiques de la chimie radicalaire. On désignera par catalyse redox ce type particulier de catalyse.L’importance de la catalyse homogène dans l’industrie chimique est difficile à déterminer parce que ses domaines d’application sont en constante évolution. Ainsi, la catalyse acido-basique voit son importance diminuer, alors que les catalyseurs redox et de coordination prennent parfois le relais des catalyseurs hétérogènes. L’introduction de catalyseurs immobilisés sur un support contribue aussi à cette difficulté et il serait préférable de considérer dans cet article la catalyse moléculaire par opposition – schématique – à la catalyse hétérogène. La nature moléculaire de ces catalyseurs autorise leur emploi dans des conditions plus douces et favorise des transformations plus efficaces. De ce fait, ils sont générateurs d’économies d’énergie et de matières premières, facteurs dont il faudra de plus en plus tenir compte.1. Catalyse acido-basiqueLes acides et les bases sont les plus simples des catalyseurs de la phase liquide et les plus anciennement connus. Plusieurs définitions leur sont associées. Selon Brønsted, un acide est une substance qui a tendance à perdre un proton; une base est une substance capable de capter un proton. La définition de Lewis englobe les acides de Brønsted ainsi que tous les composés pouvant donner ou accepter des paires d’électrons [cf. ACIDES ET BASES].Dans les réactions de catalyse acido-basique, l’une au moins des étapes est un transfert de proton du catalyseur au substrat S (catalyse acide) ou du substrat au catalyseur (catalyse basique). Le substrat intervient, de ce fait, soit comme base, soit comme acide, et la réaction implique donc un équilibre protolytique. Plusieurs possibilités peuvent apparaître. En catalyse acide:– l’équilibre protolytique est l’étape lente du processus: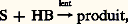 et il y a catalyse générale du premier type, où tous les acides présents peuvent intervenir;– l’équilibre protolytique n’est pas l’étape lente du processus:
et il y a catalyse générale du premier type, où tous les acides présents peuvent intervenir;– l’équilibre protolytique n’est pas l’étape lente du processus: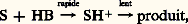 et il y a catalyse générale du deuxième type, qui est donc une catalyse générale de la deuxième étape par les bases conjuguées.Les mêmes types de réactions sont observés en catalyse basique.Les catalyseurs acides sont employés dans les réactions mettant en jeu des substrats insaturés (isomérisation, alcoylation, oligomérisation, hydratation), des acides carboxyliques et des alcools (estérification), des esters (hydrolyse), voire des substrats saturés (isomérisation d’alcanes en milieu superacide). Dans l’estérification, la première étape est la formation de l’acide conjugué de l’alcool ROH. Cet acide conjugué réagit avec l’acide carboxylique R COOH pour fournir l’ester. La réaction correspond donc à une catalyse spécifique et sa vitesse variera avec la valeur H0 du catalyseur acide. Toutefois, afin d’éviter l’hydrolyse de l’ester, l’eau, ou l’ester formé au cours de la réaction, devra être éliminé par distillation afin d’obtenir une conversion de l’acide carboxylique (soit le pourcentage en moles de cette matière première transformée) voisine de 100 p. 100.L’hydratation de l’éthylène conduit à l’éthanol: elle a lieu en présence d’acides de Brønsted comme l’acide sulfurique.Le mécanisme de cette réaction de catalyse générale acide fait intervenir la formation lente de l’ion carbénium C2H5+, qui réagit avec l’eau.L’alcoylation du benzène par différentes oléfines fournit des alkylbenzènes: elle a lieu en présence d’acides de Lewis comme le chlorure d’aluminium ou le chlorure ferrique.L’éthylbenzène, précurseur du styrène, est obtenu par réaction avec l’éthylène; le cumène, par réaction avec le propylène. La sélectivité en ces produits de réaction (soit le pourcentage en moles de ce produit de réaction par rapport à l’une des matières premières transformées) n’est pas élevée puisque des dialkyl- et des polyalkylbenzènes sont formés.Les catalyseurs basiques ont essentiellement une seule grande application industrielle, la saponification [cf. DÉTERGENTS]. Dans cette réaction, les corps gras naturels sont convertis en savons au moyen de soude caustique, ou d’autres bases, en solution aqueuse avec la libération simultanée de glycérol. Le rôle de la base est d’accélérer l’hydrolyse de l’ester selon un mécanisme de catalyse spécifique, mais la formation de l’acide gras s’accompagne de la consommation du catalyseur dans la réaction de saponification proprement dite.2. Catalyse redoxLe phénomène fondamental de cette catalyse est le fait que le transfert direct d’électrons d’un composé riche en électrons, le réducteur, à un composé pauvre en électrons, l’oxydant, est plus lent que le transfert d’électrons du réducteur au catalyseur, suivi du transfert du catalyseur à l’oxydant. Ainsi, l’ion Cu2+ catalyse l’oxydation du palladium métallique par l’oxygène moléculaire:
et il y a catalyse générale du deuxième type, qui est donc une catalyse générale de la deuxième étape par les bases conjuguées.Les mêmes types de réactions sont observés en catalyse basique.Les catalyseurs acides sont employés dans les réactions mettant en jeu des substrats insaturés (isomérisation, alcoylation, oligomérisation, hydratation), des acides carboxyliques et des alcools (estérification), des esters (hydrolyse), voire des substrats saturés (isomérisation d’alcanes en milieu superacide). Dans l’estérification, la première étape est la formation de l’acide conjugué de l’alcool ROH. Cet acide conjugué réagit avec l’acide carboxylique R COOH pour fournir l’ester. La réaction correspond donc à une catalyse spécifique et sa vitesse variera avec la valeur H0 du catalyseur acide. Toutefois, afin d’éviter l’hydrolyse de l’ester, l’eau, ou l’ester formé au cours de la réaction, devra être éliminé par distillation afin d’obtenir une conversion de l’acide carboxylique (soit le pourcentage en moles de cette matière première transformée) voisine de 100 p. 100.L’hydratation de l’éthylène conduit à l’éthanol: elle a lieu en présence d’acides de Brønsted comme l’acide sulfurique.Le mécanisme de cette réaction de catalyse générale acide fait intervenir la formation lente de l’ion carbénium C2H5+, qui réagit avec l’eau.L’alcoylation du benzène par différentes oléfines fournit des alkylbenzènes: elle a lieu en présence d’acides de Lewis comme le chlorure d’aluminium ou le chlorure ferrique.L’éthylbenzène, précurseur du styrène, est obtenu par réaction avec l’éthylène; le cumène, par réaction avec le propylène. La sélectivité en ces produits de réaction (soit le pourcentage en moles de ce produit de réaction par rapport à l’une des matières premières transformées) n’est pas élevée puisque des dialkyl- et des polyalkylbenzènes sont formés.Les catalyseurs basiques ont essentiellement une seule grande application industrielle, la saponification [cf. DÉTERGENTS]. Dans cette réaction, les corps gras naturels sont convertis en savons au moyen de soude caustique, ou d’autres bases, en solution aqueuse avec la libération simultanée de glycérol. Le rôle de la base est d’accélérer l’hydrolyse de l’ester selon un mécanisme de catalyse spécifique, mais la formation de l’acide gras s’accompagne de la consommation du catalyseur dans la réaction de saponification proprement dite.2. Catalyse redoxLe phénomène fondamental de cette catalyse est le fait que le transfert direct d’électrons d’un composé riche en électrons, le réducteur, à un composé pauvre en électrons, l’oxydant, est plus lent que le transfert d’électrons du réducteur au catalyseur, suivi du transfert du catalyseur à l’oxydant. Ainsi, l’ion Cu2+ catalyse l’oxydation du palladium métallique par l’oxygène moléculaire: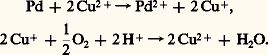 Le transfert de plusieurs électrons peut être réalisé. Il est évident que la facilité de ces transferts sera liée aux potentiels redox des ions qui interviennent. Il faut souligner que la coordination d’une molécule sur un centre métallique s’accompagne également d’un transfert d’électrons entre le métal et cette molécule devenue ligand. C’est parce qu’ils peuvent prendre divers états d’oxydation stabilisés par ces ligands que les complexes de coordination des métaux de transition facilitent les réactions de transfert d’électrons. La plupart des complexes de coordination employés en catalyse homogène sont des complexes diamagnétiques, à nombre pair d’électrons, tous appariés. Ces complexes sont le siège de processus de transferts à deux électrons; compte tenu de l’importance de ce type de catalyse, le chapitre suivant leur sera consacré. Les complexes paramagnétiques dans lesquels un électron au moins n’est pas apparié sont le siège de processus de transferts monoélectroniques et catalysent essentiellement des réactions d’oxydation radicalaires.On compte au nombre de ces réactions l’autoxydation par l’oxygène moléculaire des aldéhydes et des hydrocarbures. L’oxydation de l’éthanal, CH3CHO, est une des voies d’obtention d’acide acétique, CH3COOH. La réaction effectuée en phase liquide conduit, selon la nature de l’ion métallique employé, à l’acide acétique ou à son anhydride. L’ion métallique intervient dans l’étape d’initiation, à savoir l’étape de formation d’un radical acétyle CH3CO. par oxydation de l’éthanal:
Le transfert de plusieurs électrons peut être réalisé. Il est évident que la facilité de ces transferts sera liée aux potentiels redox des ions qui interviennent. Il faut souligner que la coordination d’une molécule sur un centre métallique s’accompagne également d’un transfert d’électrons entre le métal et cette molécule devenue ligand. C’est parce qu’ils peuvent prendre divers états d’oxydation stabilisés par ces ligands que les complexes de coordination des métaux de transition facilitent les réactions de transfert d’électrons. La plupart des complexes de coordination employés en catalyse homogène sont des complexes diamagnétiques, à nombre pair d’électrons, tous appariés. Ces complexes sont le siège de processus de transferts à deux électrons; compte tenu de l’importance de ce type de catalyse, le chapitre suivant leur sera consacré. Les complexes paramagnétiques dans lesquels un électron au moins n’est pas apparié sont le siège de processus de transferts monoélectroniques et catalysent essentiellement des réactions d’oxydation radicalaires.On compte au nombre de ces réactions l’autoxydation par l’oxygène moléculaire des aldéhydes et des hydrocarbures. L’oxydation de l’éthanal, CH3CHO, est une des voies d’obtention d’acide acétique, CH3COOH. La réaction effectuée en phase liquide conduit, selon la nature de l’ion métallique employé, à l’acide acétique ou à son anhydride. L’ion métallique intervient dans l’étape d’initiation, à savoir l’étape de formation d’un radical acétyle CH3CO. par oxydation de l’éthanal: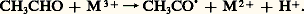 La suite de la réaction procède essentiellement selon un mécanisme radicalaire en chaîne mettant en jeu l’oxygène moléculaire.Les réactions d’autoxydation catalysées par les ions des métaux de transition sont également appliquées à l’obtention d’acide adipique (précurseur des monomères du nylon) à partir du cyclohexane, d’acide téréphtalique (un précurseur des polyesters) à partir du para-xylène.Dans tous les cas, la sélectivité en diacides n’est pas très élevée. Cette tendance est générale à toutes les réactions d’autoxydation, du fait de l’incapacité actuelle à contrôler ces réactions, même en présence de métaux de transition. Des réactions d’oxydation sélectives avec l’oxygène moléculaire interviennent cependant dans les métalloenzymes [cf. ENZYMES] et de nombreuses recherches visent à transposer ces réactions sur des systèmes moins complexes et plus appropriés aux capacités industrielles.3. Catalyse de coordinationLa catalyse de coordination concerne les processus qui mettent en œuvre des complexes des métaux de transition solubles, susceptibles d’activer, par coordination, les substrats et réactifs intervenant dans des réactions de synthèse organique. L’activation de ces partenaires en tant que ligands au sein de complexes favorise dans certaines conditions leur interaction et leur transformation en produits. La catalyse de coordination fait donc intervenir la chimie de coordination et, plus précisément, les réactions élémentaires de la chimie de coordination.Réactions élémentaires de la chimie de coordinationL’activation d’un substrat, d’un réactif s’effectue par son addition sur le centre métallique. Des sites de coordination vacants doivent donc être disponibles sur ce centre qui, rappelons-le, possède un caractère acide ou base de Lewis, ou les deux (cf. chimie de COORDINATION). Les réactions de catalyse de coordination font intervenir de ce fait des complexes précurseurs comportant au plus seize électrons dans leur couche de valence.Lorsque pratiquement un de ces caractères est présent, il y aura association d’une base (alcène, alcyne, alcadiène, monoxyde de carbone, phosphine, etc.) ou d’un acide (proton). Dans le cas des bases, le nombre d’électrons présents sur le centre métallique croît de deux unités. La réaction inverse, la dissociation , libère un site de coordination. La combinaison d’une dissociation et d’une association d’un couple ligand-substrat conduira à la substitution du ligand par ce substrat. Lorsque les deux caractères sont présents simultanément, on peut observer l’addition oxydante d’un réactif XY. La rupture de la liaison XY (hydrogène moléculaire, hydracides, halogènes, halogénures organiques) n’est pas générale dès lors que le réactif contient une liaison multiple (oxygène, composés perfluorés insaturés...). La formation de deux liaisons covalentes dans ce processus implique la présence de deux sites de coordination vacants sur le centre métallique. L’élimination réductrice , processus inverse de l’addition oxydante, conduit à la création d’une liaison carbone-carbone lorsque les fragments X et Y sont liés par un atome de carbone au métal.La présence de deux ligands différents sur le centre métallique peut conduire, du fait de leur activation, à leur couplage, ce qui donne un nouveau ligand. Le processus de déplacement-1,2 de ligands est d’une importance capitale en catalyse homogène: il est bien connu pour les réactions qui mettent en jeu les ligands hydrure et alcène, alkyle et carbonyle, alkyle et carbène et alkyle et alcène (cf. tabl. 1). Les processus de déplacement-1,2 de ligands amènent la création d’un site de coordination vacant sur le métal ainsi qu’une diminution du nombre d’électrons sur ce dernier de deux unités. Ce mécanisme, comme les précédents, possède son inverse, à savoir une régression-1,2 qui prend le nom de 廓-élimination lorsque les ligands hydrure et alcène sont formés.Enfin, la réaction de cyclométallation consiste en le couplage dans la sphère de coordination de deux ligands présentant chacun une ou plusieurs liaisons de type 神. Au cours de ce processus, également réversible, le nombre d’électrons sur le centre métallique ainsi que son état d’oxydation formel varient de deux unités. Un tel mécanisme, généralement appelé couplage ou doublement oxydant, donne lieu à la formation d’une liaison carbone-carbone dans le cas des substrats hydrocarbonés. Comme deux liaisons métal-carbone sont également formées, il représente donc un cas extrêmement intéressant au niveau réactionnel. Un processus de cyclométallation intervient aussi lors de l’interaction entre un métallocarbène et un alcène ou alcyne. Un tel mécanisme, également réversible, conduit à la formation d’un métallacyclobutane.Réactions de catalyse de coordinationEn catalyse de coordination, les réactions catalytiques sont des successions de réactions élémentaires rapides qui font intervenir différents substrats et réactifs et un précurseur catalytique (complexe de coordination) continuellement régénéré. La régénération peut être spontanée, comme dans la réaction d’hydroformylation des alcènes (fig. 1), ou le précurseur est reformé au cours de réactions ultérieures, comme dans le cas du procédé Wacker de synthèse de l’éthanal (fig. 2). La catalyse de coordination excelle dans la formation sélective de liaisons carbone-carbone et carbone-hydrogène. Elle a également rencontré quelques succès pour les réactions de formation de liaisons carbone-oxygène, mais des méthodes générales de formation d’autres liaisons, par exemple carbone-azote, font encore défaut.Réactions de formation de liaisons carbone-carboneLa condensation des hydrocarbures insaturés s’effectue avec de nombreux systèmes catalytiques, obtenus initialement, selon la technique de Ziegler, par réduction d’un sel de métal de transition soluble en milieu organique (carboxylate, acétylacétonate) par les composés organoaluminiques Rn AlCl3-n . Des systèmes plus élaborés et mieux caractérisés ont été par la suite découverts, grâce à l’étude des mécanismes réactionnels, voire à l’isolement d’intermédiaires réactionnels ou à la préparation de complexes comportant des ligands labiles facilement déplacés par ces hydrocarbures insaturés. Le tableau 2 consigne quelques exemples typiques de tels composés, qui sont en fait des précurseurs catalytiques.Selon les vitesses respectives d’incorporation du substrat via un processus de déplacement-1,2 ou de cyclométallation et d’élimination des produits de la réaction, on observera la polymérisation, l’oligomérisation, la trimérisation, la dimérisation de ce substrat. Les produits formés ont des applications multiples dans les domaines:– des matières plastiques: polyéthylène, polypropylène, terpolymères éthylène-propylène-diènes, polybutadiène et matières premières pour l’obtention de terpolymères (hexa-1,4-diène par codimérisation du butadiène et de l’éthylène);– des fibres synthétiques: matières premières (cyclooctadiène, cyclododécatriène) pour l’obtention de diacides, de diamines, de lactames entrant dans la préparation de polyesters et de polyamides;– des plastifiants: matières premières pour la synthèse d’esters;– des additifs de carburant: matières premières pour la préparation d’hydrocarbures à haut indice d’octane (diméthylbutane par dimérisation du propylène et hydrogénation);– des parfums: matières premières pour la synthèse totale de produits naturels (dimères du butadiène et de l’isoprène) ou nouveaux (télomères du butadiène et de l’isoprène).Les études des mécanismes de ces réactions, quoique très nombreuses, n’ont pu éclaircir totalement les modes d’action de ces catalyseurs. Toutefois, des schémas qualitatifs ont été établis, qui démontrent l’importance sur le cours de la réaction des ligands autres que les substrats et les réactifs présents dans la sphère de coordination de l’espèce métallique. De par leur aptitude à donner ou à accepter des électrons, ces ligands ancillaires influencent les propriétés électroniques du centre métallique: de par leur volume parfois important, ils modifient les propriétés stériques du centre métallique. Un accroissement de la vitesse de réaction tout comme une augmentation de la sélectivité de la réaction pour le ou les produits souhaités sont de fait accessibles par la modification «à façon» du catalyseur.Certaines de ces réactions mettent en jeu des métallocarbènes et des métallocarbynes. On observe ainsi des réactions de redistribution des groupements alkylidène et alkylidyne, constituants respectifs des alcènes et des alcynes lors de la métathèse de ces substrats. La métathèse du stilbène et de l’éthylène conduit, par exemple, à la formation de deux molécules de styrène (tabl. 2). La polymérisation par ouverture de cycle du cyclopentène, qui fournit des polyalcénamères, est une réaction de métathèse. La métathèse d’alcènes fonctionnalisés, comme certains acides gras naturels, présente l’intérêt de fournir des composés comportant une fonction à chaque extrémité de chaîne.Lorsqu’un des partenaires réactionnels dans les réactions d’oligomérisation est remplacé par le monoxyde de carbone, une réaction de carbonylation de la liaison métal-carbone peut intervenir. Cette nouvelle réaction présente l’intérêt d’introduire une fonction oxygénée dans le(s) produit(s) réactionnel(s) après coupure de la liaison métal-acyle. Cette coupure peut être effectuée par l’hydrogène moléculaire ou par un composé protonique ZH. Dans les deux cas, une espèce métal hydrure est obtenue, espèce qui pourra réagir avec un substrat insaturé pour reconduire à une liaison métal-carbone. La juxtaposition de ces différents processus dans un ordre privilégié démontre les possibilités de la catalyse de coordination. Ainsi, dans l’hydroformylation des alcènes (fig. 1), trois molécules différentes interagissent pour conduire spécifiquement à des aldéhydes. Les réactions de carbonylation sont d’une grande importance pour l’obtention, à l’échelle industrielle, d’aldéhydes, d’alcools et d’acides carboxyliques. Le butanal, obtenu par hydroformylation du propylène, est la matière première de la synthèse de l’éthyl-2-hexanol employé dans l’industrie des plastifiants de polymères tels que le chlorure de polyvinyle. L’acide acétique, utilisé pour la préparation d’esters employés comme solvants ou intermédiaires de synthèse (acétate de vinyle), est obtenu avec une sélectivité très élevée par carbonylation du méthanol (procédé Monsanto, tabl. 2). L’insertion du monoxyde de carbone dans une liaison métal-hydrogène, bien qu’elle soit plus difficile à réaliser que dans une liaison métal-carbone, ouvre de nouvelles voies de synthèse à partir de molécules aussi simples que le monoxyde de carbone et que l’hydrogène; l’éthylène glycol, HOCH2CH2OH, composant essentiel des liquides de refroidissement des moteurs à explosion, peut être obtenu à partir de ces éléments, les agrégats moléculaires (par exemple Ru3(CO)12, Rh6(CO)16 associés à différents types de bases) ainsi que des pressions élevées (supérieures à 50 MPa).Dans quelques cas, le rôle du centre métallique consiste seulement en l’activation du substrat afin d’exalter sa réactivité vis-à-vis d’un réactif électrophile ou nucléophile. L’obtention d’adiponitrile à partir de butadiène et d’acide cyanhydrique met en œuvre deux réactions consécutives d’attaque nucléophile de l’anion cyanure, le nucléophile, sur les ligands dérivés du butadiène liés à un ion nickel (II): successivement un ligand allylique puis un ligand alcène (procédé DuPont-Rhône Poulenc).Réactions de formation de liaisons carbone-hydrogènePratiquement tous les métaux de transition sont actifs en catalyse d’hydrogénation. L’hydrogénation des alcènes, alcynes, alcadiènes-1,3, des hétéro-doubles et triples liaisons a été décrite. À nouveau, l’étape essentielle de cette réaction est un processus de déplacement-1,2 qui met en œuvre un ligand hydrogène et le substrat insaturé. La figure 3 présente un des cycles catalytiques envisageables pour des complexes du rhodium associé à des phosphines symbolisées par L. Une des applications les plus élégantes de la catalyse de coordination est la synthèse de la L-dihydroxy-3,4-phénylalanine (L-DOPA) qui est utilisée dans le traitement de la maladie de Parkinson. L’étape clé de cette synthèse est l’hydrogénation spécifique d’un acide acétamidocinnamique en un dérivé de la L-DOPA. L’induction asymétrique au cours de l’hydrogénation est effectuée par emploi d’un complexe du rhodium qui comporte une phosphine optiquement active.Réactions de formation de liaisons carbone-oxygèneLes réactions de formation de liaisons C-O catalysées par des complexes de métaux de transition sont peu nombreuses, mais revêtent un grand intérêt industriel à cause de l’importance des produits formés. Les réactions mettant en jeu directement l’oxygène moléculaire sont, nous l’avons vu, peu sélectives. De ce fait, ce sont des réactifs contenant au moins un atome d’oxygène qui sont employés dans les réactions hautement sélectives de formation de liaisons C-O.Lorsqu’un réactif nucléophile est employé, par exemple l’eau, la formation d’éthanal, dans le cas de l’éthylène, et de cétones, dans le cas des autres alcènes, est observée en présence de sels de palladium (II). Toutefois (fig. 2), le palladium est le composé oxydant et se trouve réduit à l’état métallique en fin de réaction. La réoxydation du palladium est assurée par des ions Cu2+ en présence d’oxygène moléculaire, selon une catalyse redox.Lorsqu’un réactif électrophile est employé, par exemple un hydroperoxyde, la formation d’époxyde est observée en présence de composés du molybdène, du vanadium ou du titane. La réaction est principalement employée pour la synthèse, à partir d’hydroperoxyde de t -butyle et de propylène, d’époxypropane qui est un intermédiaire dans la préparation de propylène glycol et de polyéthers (procédé Halcon). L’époxydation des alcools allyliques prochiraux en présence d’un catalyseur optiquement actif contenant, par exemple, un ligand dérivé de l’acide tartrique, permet d’accéder à des composés chiraux avec une induction asymétrique totale .4. Catalyseurs hybridesParmi les problèmes de la catalyse homogène, la séparation du catalyseur des produits de la réaction ainsi que leur récupération intégrale lorsque des composés des métaux précieux sont employés sont importantes, et différentes techniques se sont employées à les résoudre. Parfois, une simple distillation suffit (procédé Wacker), mais il faut souvent un cycle complexe de récupération du catalyseur (hydroformylation des alcènes au contact de complexes du cobalt). C’est pourquoi le concept d’opérer en un système à deux phases, voire trois, a été introduit voici quelques années et devrait donner lieu à des applications de plus en plus nombreuses. Un premier développement de ce concept consiste à effectuer la réaction dans un milieu constitué de deux solvants non miscibles. Le catalyseur est préférentiellement soluble dans une de ces phases, les produits de la réaction dans l’autre, et la réaction catalytique s’effectue. L’emploi de phosphines solubles dans l’eau et par conséquent de catalyseurs solubles dans ce milieu a ainsi été préconisé.Un second développement de ce concept conduit à l’utilisation d’un solide jouant le rôle soit de réservoir du catalyseur moléculaire, soit de support de ce catalyseur qui est alors chimiquement lié (greffé) à la surface du solide.Dans le premier cas, la réaction catalytique a lieu en solution; après réaction, les produits sont distillés, et, au cours de ce traitement, le catalyseur, instable, se dépose sur le solide qui peut être un charbon, un oxyde amorphe ou cristallisé. Des variantes du procédé Wacker utilisent ainsi le palladium déposé sur charbon; l’hydroformylation de certains alcènes est effectuée grâce à des catalyseurs à base de rhodium déposé sur alumine; des résines sulfonées, des alumines imprégnées de chlorure d’aluminium sont employées en catalyse acide.Dans le second cas, la réaction catalytique a lieu à la surface du support mais procède d’un mécanisme moléculaire qui fait intervenir le greffon moléculaire. Ces catalyseurs sont dits hybrides, immobilisés ou encore supportés. Les supports peuvent être des solides inorganiques (oxydes, charbons, argiles, etc.) ou des polymères organiques (polystyrènes, polyamides, copolymères, etc.) qui sont modifiés en conséquence pour former des liaisons chimiques avec des catalyseurs moléculaires. La séparation des centres actifs lors du greffage peut augmenter la durée de vie de l’espèce catalytique en l’empêchant de se recombiner ou de réagir sur des impuretés accompagnant les partenaires réactionnels ou les produits de réaction. De plus, le support se comporte comme un ligand qui pourra également influer sur la sélectivité de la réaction. Enfin, ces catalyseurs, parce qu’ils sont facilement éliminés du milieu réactionnel, permettent l’obtention de produits d’une plus grande pureté.Les catalyseurs hybrides sont d’introduction encore trop récente pour que l’on puisse évaluer leur impact industriel. Leurs applications essentielles résident dans le domaine de la polymérisation des alcènes (éthylène, propylène). Toutefois, la possibilité de combiner les aspects positifs de la catalyse hétérogène et de la catalyse homogène, notamment de coordination, ouvre des perspectives intéressantes. On peut concevoir des catalyseurs remplissant plusieurs fonctions, mises en œuvre de manière séquentielle, des catalyseurs associant une catalyse hétérogène avec une catalyse de coordination, ou encore des catalyseurs mimant les systèmes enzymatiques. Enfin, il faut souligner que l’association de composés moléculaires bien définis et de supports peut conduire, dans des conditions contrôlées, à la formation de catalyseurs hétérogènes. Les systèmes présentent alors des propriétés supérieures en catalyse de contact.5. Aspects industriels et économiquesUn procédé de synthèse chimique peut être représenté schématiquement par la séquence:
La suite de la réaction procède essentiellement selon un mécanisme radicalaire en chaîne mettant en jeu l’oxygène moléculaire.Les réactions d’autoxydation catalysées par les ions des métaux de transition sont également appliquées à l’obtention d’acide adipique (précurseur des monomères du nylon) à partir du cyclohexane, d’acide téréphtalique (un précurseur des polyesters) à partir du para-xylène.Dans tous les cas, la sélectivité en diacides n’est pas très élevée. Cette tendance est générale à toutes les réactions d’autoxydation, du fait de l’incapacité actuelle à contrôler ces réactions, même en présence de métaux de transition. Des réactions d’oxydation sélectives avec l’oxygène moléculaire interviennent cependant dans les métalloenzymes [cf. ENZYMES] et de nombreuses recherches visent à transposer ces réactions sur des systèmes moins complexes et plus appropriés aux capacités industrielles.3. Catalyse de coordinationLa catalyse de coordination concerne les processus qui mettent en œuvre des complexes des métaux de transition solubles, susceptibles d’activer, par coordination, les substrats et réactifs intervenant dans des réactions de synthèse organique. L’activation de ces partenaires en tant que ligands au sein de complexes favorise dans certaines conditions leur interaction et leur transformation en produits. La catalyse de coordination fait donc intervenir la chimie de coordination et, plus précisément, les réactions élémentaires de la chimie de coordination.Réactions élémentaires de la chimie de coordinationL’activation d’un substrat, d’un réactif s’effectue par son addition sur le centre métallique. Des sites de coordination vacants doivent donc être disponibles sur ce centre qui, rappelons-le, possède un caractère acide ou base de Lewis, ou les deux (cf. chimie de COORDINATION). Les réactions de catalyse de coordination font intervenir de ce fait des complexes précurseurs comportant au plus seize électrons dans leur couche de valence.Lorsque pratiquement un de ces caractères est présent, il y aura association d’une base (alcène, alcyne, alcadiène, monoxyde de carbone, phosphine, etc.) ou d’un acide (proton). Dans le cas des bases, le nombre d’électrons présents sur le centre métallique croît de deux unités. La réaction inverse, la dissociation , libère un site de coordination. La combinaison d’une dissociation et d’une association d’un couple ligand-substrat conduira à la substitution du ligand par ce substrat. Lorsque les deux caractères sont présents simultanément, on peut observer l’addition oxydante d’un réactif XY. La rupture de la liaison XY (hydrogène moléculaire, hydracides, halogènes, halogénures organiques) n’est pas générale dès lors que le réactif contient une liaison multiple (oxygène, composés perfluorés insaturés...). La formation de deux liaisons covalentes dans ce processus implique la présence de deux sites de coordination vacants sur le centre métallique. L’élimination réductrice , processus inverse de l’addition oxydante, conduit à la création d’une liaison carbone-carbone lorsque les fragments X et Y sont liés par un atome de carbone au métal.La présence de deux ligands différents sur le centre métallique peut conduire, du fait de leur activation, à leur couplage, ce qui donne un nouveau ligand. Le processus de déplacement-1,2 de ligands est d’une importance capitale en catalyse homogène: il est bien connu pour les réactions qui mettent en jeu les ligands hydrure et alcène, alkyle et carbonyle, alkyle et carbène et alkyle et alcène (cf. tabl. 1). Les processus de déplacement-1,2 de ligands amènent la création d’un site de coordination vacant sur le métal ainsi qu’une diminution du nombre d’électrons sur ce dernier de deux unités. Ce mécanisme, comme les précédents, possède son inverse, à savoir une régression-1,2 qui prend le nom de 廓-élimination lorsque les ligands hydrure et alcène sont formés.Enfin, la réaction de cyclométallation consiste en le couplage dans la sphère de coordination de deux ligands présentant chacun une ou plusieurs liaisons de type 神. Au cours de ce processus, également réversible, le nombre d’électrons sur le centre métallique ainsi que son état d’oxydation formel varient de deux unités. Un tel mécanisme, généralement appelé couplage ou doublement oxydant, donne lieu à la formation d’une liaison carbone-carbone dans le cas des substrats hydrocarbonés. Comme deux liaisons métal-carbone sont également formées, il représente donc un cas extrêmement intéressant au niveau réactionnel. Un processus de cyclométallation intervient aussi lors de l’interaction entre un métallocarbène et un alcène ou alcyne. Un tel mécanisme, également réversible, conduit à la formation d’un métallacyclobutane.Réactions de catalyse de coordinationEn catalyse de coordination, les réactions catalytiques sont des successions de réactions élémentaires rapides qui font intervenir différents substrats et réactifs et un précurseur catalytique (complexe de coordination) continuellement régénéré. La régénération peut être spontanée, comme dans la réaction d’hydroformylation des alcènes (fig. 1), ou le précurseur est reformé au cours de réactions ultérieures, comme dans le cas du procédé Wacker de synthèse de l’éthanal (fig. 2). La catalyse de coordination excelle dans la formation sélective de liaisons carbone-carbone et carbone-hydrogène. Elle a également rencontré quelques succès pour les réactions de formation de liaisons carbone-oxygène, mais des méthodes générales de formation d’autres liaisons, par exemple carbone-azote, font encore défaut.Réactions de formation de liaisons carbone-carboneLa condensation des hydrocarbures insaturés s’effectue avec de nombreux systèmes catalytiques, obtenus initialement, selon la technique de Ziegler, par réduction d’un sel de métal de transition soluble en milieu organique (carboxylate, acétylacétonate) par les composés organoaluminiques Rn AlCl3-n . Des systèmes plus élaborés et mieux caractérisés ont été par la suite découverts, grâce à l’étude des mécanismes réactionnels, voire à l’isolement d’intermédiaires réactionnels ou à la préparation de complexes comportant des ligands labiles facilement déplacés par ces hydrocarbures insaturés. Le tableau 2 consigne quelques exemples typiques de tels composés, qui sont en fait des précurseurs catalytiques.Selon les vitesses respectives d’incorporation du substrat via un processus de déplacement-1,2 ou de cyclométallation et d’élimination des produits de la réaction, on observera la polymérisation, l’oligomérisation, la trimérisation, la dimérisation de ce substrat. Les produits formés ont des applications multiples dans les domaines:– des matières plastiques: polyéthylène, polypropylène, terpolymères éthylène-propylène-diènes, polybutadiène et matières premières pour l’obtention de terpolymères (hexa-1,4-diène par codimérisation du butadiène et de l’éthylène);– des fibres synthétiques: matières premières (cyclooctadiène, cyclododécatriène) pour l’obtention de diacides, de diamines, de lactames entrant dans la préparation de polyesters et de polyamides;– des plastifiants: matières premières pour la synthèse d’esters;– des additifs de carburant: matières premières pour la préparation d’hydrocarbures à haut indice d’octane (diméthylbutane par dimérisation du propylène et hydrogénation);– des parfums: matières premières pour la synthèse totale de produits naturels (dimères du butadiène et de l’isoprène) ou nouveaux (télomères du butadiène et de l’isoprène).Les études des mécanismes de ces réactions, quoique très nombreuses, n’ont pu éclaircir totalement les modes d’action de ces catalyseurs. Toutefois, des schémas qualitatifs ont été établis, qui démontrent l’importance sur le cours de la réaction des ligands autres que les substrats et les réactifs présents dans la sphère de coordination de l’espèce métallique. De par leur aptitude à donner ou à accepter des électrons, ces ligands ancillaires influencent les propriétés électroniques du centre métallique: de par leur volume parfois important, ils modifient les propriétés stériques du centre métallique. Un accroissement de la vitesse de réaction tout comme une augmentation de la sélectivité de la réaction pour le ou les produits souhaités sont de fait accessibles par la modification «à façon» du catalyseur.Certaines de ces réactions mettent en jeu des métallocarbènes et des métallocarbynes. On observe ainsi des réactions de redistribution des groupements alkylidène et alkylidyne, constituants respectifs des alcènes et des alcynes lors de la métathèse de ces substrats. La métathèse du stilbène et de l’éthylène conduit, par exemple, à la formation de deux molécules de styrène (tabl. 2). La polymérisation par ouverture de cycle du cyclopentène, qui fournit des polyalcénamères, est une réaction de métathèse. La métathèse d’alcènes fonctionnalisés, comme certains acides gras naturels, présente l’intérêt de fournir des composés comportant une fonction à chaque extrémité de chaîne.Lorsqu’un des partenaires réactionnels dans les réactions d’oligomérisation est remplacé par le monoxyde de carbone, une réaction de carbonylation de la liaison métal-carbone peut intervenir. Cette nouvelle réaction présente l’intérêt d’introduire une fonction oxygénée dans le(s) produit(s) réactionnel(s) après coupure de la liaison métal-acyle. Cette coupure peut être effectuée par l’hydrogène moléculaire ou par un composé protonique ZH. Dans les deux cas, une espèce métal hydrure est obtenue, espèce qui pourra réagir avec un substrat insaturé pour reconduire à une liaison métal-carbone. La juxtaposition de ces différents processus dans un ordre privilégié démontre les possibilités de la catalyse de coordination. Ainsi, dans l’hydroformylation des alcènes (fig. 1), trois molécules différentes interagissent pour conduire spécifiquement à des aldéhydes. Les réactions de carbonylation sont d’une grande importance pour l’obtention, à l’échelle industrielle, d’aldéhydes, d’alcools et d’acides carboxyliques. Le butanal, obtenu par hydroformylation du propylène, est la matière première de la synthèse de l’éthyl-2-hexanol employé dans l’industrie des plastifiants de polymères tels que le chlorure de polyvinyle. L’acide acétique, utilisé pour la préparation d’esters employés comme solvants ou intermédiaires de synthèse (acétate de vinyle), est obtenu avec une sélectivité très élevée par carbonylation du méthanol (procédé Monsanto, tabl. 2). L’insertion du monoxyde de carbone dans une liaison métal-hydrogène, bien qu’elle soit plus difficile à réaliser que dans une liaison métal-carbone, ouvre de nouvelles voies de synthèse à partir de molécules aussi simples que le monoxyde de carbone et que l’hydrogène; l’éthylène glycol, HOCH2CH2OH, composant essentiel des liquides de refroidissement des moteurs à explosion, peut être obtenu à partir de ces éléments, les agrégats moléculaires (par exemple Ru3(CO)12, Rh6(CO)16 associés à différents types de bases) ainsi que des pressions élevées (supérieures à 50 MPa).Dans quelques cas, le rôle du centre métallique consiste seulement en l’activation du substrat afin d’exalter sa réactivité vis-à-vis d’un réactif électrophile ou nucléophile. L’obtention d’adiponitrile à partir de butadiène et d’acide cyanhydrique met en œuvre deux réactions consécutives d’attaque nucléophile de l’anion cyanure, le nucléophile, sur les ligands dérivés du butadiène liés à un ion nickel (II): successivement un ligand allylique puis un ligand alcène (procédé DuPont-Rhône Poulenc).Réactions de formation de liaisons carbone-hydrogènePratiquement tous les métaux de transition sont actifs en catalyse d’hydrogénation. L’hydrogénation des alcènes, alcynes, alcadiènes-1,3, des hétéro-doubles et triples liaisons a été décrite. À nouveau, l’étape essentielle de cette réaction est un processus de déplacement-1,2 qui met en œuvre un ligand hydrogène et le substrat insaturé. La figure 3 présente un des cycles catalytiques envisageables pour des complexes du rhodium associé à des phosphines symbolisées par L. Une des applications les plus élégantes de la catalyse de coordination est la synthèse de la L-dihydroxy-3,4-phénylalanine (L-DOPA) qui est utilisée dans le traitement de la maladie de Parkinson. L’étape clé de cette synthèse est l’hydrogénation spécifique d’un acide acétamidocinnamique en un dérivé de la L-DOPA. L’induction asymétrique au cours de l’hydrogénation est effectuée par emploi d’un complexe du rhodium qui comporte une phosphine optiquement active.Réactions de formation de liaisons carbone-oxygèneLes réactions de formation de liaisons C-O catalysées par des complexes de métaux de transition sont peu nombreuses, mais revêtent un grand intérêt industriel à cause de l’importance des produits formés. Les réactions mettant en jeu directement l’oxygène moléculaire sont, nous l’avons vu, peu sélectives. De ce fait, ce sont des réactifs contenant au moins un atome d’oxygène qui sont employés dans les réactions hautement sélectives de formation de liaisons C-O.Lorsqu’un réactif nucléophile est employé, par exemple l’eau, la formation d’éthanal, dans le cas de l’éthylène, et de cétones, dans le cas des autres alcènes, est observée en présence de sels de palladium (II). Toutefois (fig. 2), le palladium est le composé oxydant et se trouve réduit à l’état métallique en fin de réaction. La réoxydation du palladium est assurée par des ions Cu2+ en présence d’oxygène moléculaire, selon une catalyse redox.Lorsqu’un réactif électrophile est employé, par exemple un hydroperoxyde, la formation d’époxyde est observée en présence de composés du molybdène, du vanadium ou du titane. La réaction est principalement employée pour la synthèse, à partir d’hydroperoxyde de t -butyle et de propylène, d’époxypropane qui est un intermédiaire dans la préparation de propylène glycol et de polyéthers (procédé Halcon). L’époxydation des alcools allyliques prochiraux en présence d’un catalyseur optiquement actif contenant, par exemple, un ligand dérivé de l’acide tartrique, permet d’accéder à des composés chiraux avec une induction asymétrique totale .4. Catalyseurs hybridesParmi les problèmes de la catalyse homogène, la séparation du catalyseur des produits de la réaction ainsi que leur récupération intégrale lorsque des composés des métaux précieux sont employés sont importantes, et différentes techniques se sont employées à les résoudre. Parfois, une simple distillation suffit (procédé Wacker), mais il faut souvent un cycle complexe de récupération du catalyseur (hydroformylation des alcènes au contact de complexes du cobalt). C’est pourquoi le concept d’opérer en un système à deux phases, voire trois, a été introduit voici quelques années et devrait donner lieu à des applications de plus en plus nombreuses. Un premier développement de ce concept consiste à effectuer la réaction dans un milieu constitué de deux solvants non miscibles. Le catalyseur est préférentiellement soluble dans une de ces phases, les produits de la réaction dans l’autre, et la réaction catalytique s’effectue. L’emploi de phosphines solubles dans l’eau et par conséquent de catalyseurs solubles dans ce milieu a ainsi été préconisé.Un second développement de ce concept conduit à l’utilisation d’un solide jouant le rôle soit de réservoir du catalyseur moléculaire, soit de support de ce catalyseur qui est alors chimiquement lié (greffé) à la surface du solide.Dans le premier cas, la réaction catalytique a lieu en solution; après réaction, les produits sont distillés, et, au cours de ce traitement, le catalyseur, instable, se dépose sur le solide qui peut être un charbon, un oxyde amorphe ou cristallisé. Des variantes du procédé Wacker utilisent ainsi le palladium déposé sur charbon; l’hydroformylation de certains alcènes est effectuée grâce à des catalyseurs à base de rhodium déposé sur alumine; des résines sulfonées, des alumines imprégnées de chlorure d’aluminium sont employées en catalyse acide.Dans le second cas, la réaction catalytique a lieu à la surface du support mais procède d’un mécanisme moléculaire qui fait intervenir le greffon moléculaire. Ces catalyseurs sont dits hybrides, immobilisés ou encore supportés. Les supports peuvent être des solides inorganiques (oxydes, charbons, argiles, etc.) ou des polymères organiques (polystyrènes, polyamides, copolymères, etc.) qui sont modifiés en conséquence pour former des liaisons chimiques avec des catalyseurs moléculaires. La séparation des centres actifs lors du greffage peut augmenter la durée de vie de l’espèce catalytique en l’empêchant de se recombiner ou de réagir sur des impuretés accompagnant les partenaires réactionnels ou les produits de réaction. De plus, le support se comporte comme un ligand qui pourra également influer sur la sélectivité de la réaction. Enfin, ces catalyseurs, parce qu’ils sont facilement éliminés du milieu réactionnel, permettent l’obtention de produits d’une plus grande pureté.Les catalyseurs hybrides sont d’introduction encore trop récente pour que l’on puisse évaluer leur impact industriel. Leurs applications essentielles résident dans le domaine de la polymérisation des alcènes (éthylène, propylène). Toutefois, la possibilité de combiner les aspects positifs de la catalyse hétérogène et de la catalyse homogène, notamment de coordination, ouvre des perspectives intéressantes. On peut concevoir des catalyseurs remplissant plusieurs fonctions, mises en œuvre de manière séquentielle, des catalyseurs associant une catalyse hétérogène avec une catalyse de coordination, ou encore des catalyseurs mimant les systèmes enzymatiques. Enfin, il faut souligner que l’association de composés moléculaires bien définis et de supports peut conduire, dans des conditions contrôlées, à la formation de catalyseurs hétérogènes. Les systèmes présentent alors des propriétés supérieures en catalyse de contact.5. Aspects industriels et économiquesUn procédé de synthèse chimique peut être représenté schématiquement par la séquence: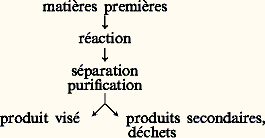 Un bon procédé nécessite une haute efficacité dans chacune de ses étapes. En outre, l’industrie chimique contemporaine doit faire face à des contraintes imposées par les facteurs économiques et les dispositions législatives. Parmi ces contraintes, on citera l’utilisation d’énergie, la valorisation ou l’emploi de produits secondaires, le choix des matières premières, le contrôle de la pollution.Toute réaction chimique demande de l’énergie pour se faire et, dans la conjoncture économique actuelle, il convient donc de rechercher le procédé à faible consommation d’énergie (procédés autothermiques, à basses pressions, à faibles températures). On observe cette tendance dans le domaine de l’hydroformylation où le remplacement des complexes du cobalt par des complexes du rhodium a amené une diminution de la pression et de la température de réaction, a amélioré la sélectivité et a supprimé le cycle de retraitement du catalyseur.Toute séparation ou purification requiert également de l’énergie et peut être parfois plus onéreuse que la mise en œuvre de la réaction elle-même. En outre, l’obtention de produits secondaires et de déchets se fait aux dépens, non seulement du coût global du procédé, mais aussi de la perte de matières premières. La nécessité de supprimer ces sous-produits exige la recherche de réactions de haute spécificité. Toutefois, lorsque la nature même du procédé l’interdit, les sous-produits doivent pouvoir être valorisés dans d’autres réactions. C’est le cas du procédé Halcon, générateur d’alcool t -butylique qui est utilisé comme matière première dans la synthèse du caoutchouc butyl ou comme additif au carburant auto: les deux procédés sont de ce fait couplés sur le même site industriel.Le grand attrait de la catalyse de coordination est le degré élevé de sélectivité atteint dans des réactions qui peuvent donner plusieurs produits possibles. La synthèse de la L-DOPA en est un magnifique exemple. Il ne fait pas de doute que ce type de catalyse trouvera un vaste champ d’applications dans la préparation de produits à haute valeur ajoutée comme les médicaments, les parfums, les flaveurs, les pesticides: de nombreuses publications scientifiques présentent déjà de telles possibilités. Néanmoins, les complexes de coordination interviennent dans de nombreux procédés de l’industrie chimique lourde et sont mis en œuvre dans des unités de production de 10 000 à 100 000 t/an.L’utilisation croissante des métaux précieux (rhodium, palladium, iridium, platine) démontre que le coût global du procédé ne fait pas intervenir de manière rédhibitoire l’investissement en ces métaux mais dépend davantage des investissements techniques. Les coûts différenciés des matières premières, dépendant fortement de la demande sur les marchés, amèneront de plus en plus une compétition entre procédés homogènes (voire hybrides) et hétérogènes au moment de leur réalisation industrielle. Ainsi, le styrène pourrait être obtenu par des procédés de catalyse homogène mettant en œuvre d’autres matières premières. On peut également envisager le remplacement à long terme du gaz naturel et du pétrole comme sources de matières premières de l’industrie chimique organique. L’utilisation d’un mélange d’hydrogène et de monoxyde de carbone (gaz de synthèse) apparaît comme une solution raisonnable: la production d’acide acétique par carbonylation du méthanol, lui-même obtenu à partir du gaz de synthèse, pourrait ainsi se substituer à l’oxydation de l’éthanal dont la synthèse s’effectue selon le procédé Wacker. Plusieurs grandes nations industrialisées ont déjà défini des programmes de recherche et de développement ambitieux sur la carbochimie et des applications sont déjà en cours de réalisation. Ainsi, dès 1983, une usine américaine a produit de l’anhydride acétique par une voie purement carbochimique qui met en œuvre un catalyseur hétérogène pour la synthèse du méthanol et un catalyseur homogène pour sa transformation en anhydride acétique.
Un bon procédé nécessite une haute efficacité dans chacune de ses étapes. En outre, l’industrie chimique contemporaine doit faire face à des contraintes imposées par les facteurs économiques et les dispositions législatives. Parmi ces contraintes, on citera l’utilisation d’énergie, la valorisation ou l’emploi de produits secondaires, le choix des matières premières, le contrôle de la pollution.Toute réaction chimique demande de l’énergie pour se faire et, dans la conjoncture économique actuelle, il convient donc de rechercher le procédé à faible consommation d’énergie (procédés autothermiques, à basses pressions, à faibles températures). On observe cette tendance dans le domaine de l’hydroformylation où le remplacement des complexes du cobalt par des complexes du rhodium a amené une diminution de la pression et de la température de réaction, a amélioré la sélectivité et a supprimé le cycle de retraitement du catalyseur.Toute séparation ou purification requiert également de l’énergie et peut être parfois plus onéreuse que la mise en œuvre de la réaction elle-même. En outre, l’obtention de produits secondaires et de déchets se fait aux dépens, non seulement du coût global du procédé, mais aussi de la perte de matières premières. La nécessité de supprimer ces sous-produits exige la recherche de réactions de haute spécificité. Toutefois, lorsque la nature même du procédé l’interdit, les sous-produits doivent pouvoir être valorisés dans d’autres réactions. C’est le cas du procédé Halcon, générateur d’alcool t -butylique qui est utilisé comme matière première dans la synthèse du caoutchouc butyl ou comme additif au carburant auto: les deux procédés sont de ce fait couplés sur le même site industriel.Le grand attrait de la catalyse de coordination est le degré élevé de sélectivité atteint dans des réactions qui peuvent donner plusieurs produits possibles. La synthèse de la L-DOPA en est un magnifique exemple. Il ne fait pas de doute que ce type de catalyse trouvera un vaste champ d’applications dans la préparation de produits à haute valeur ajoutée comme les médicaments, les parfums, les flaveurs, les pesticides: de nombreuses publications scientifiques présentent déjà de telles possibilités. Néanmoins, les complexes de coordination interviennent dans de nombreux procédés de l’industrie chimique lourde et sont mis en œuvre dans des unités de production de 10 000 à 100 000 t/an.L’utilisation croissante des métaux précieux (rhodium, palladium, iridium, platine) démontre que le coût global du procédé ne fait pas intervenir de manière rédhibitoire l’investissement en ces métaux mais dépend davantage des investissements techniques. Les coûts différenciés des matières premières, dépendant fortement de la demande sur les marchés, amèneront de plus en plus une compétition entre procédés homogènes (voire hybrides) et hétérogènes au moment de leur réalisation industrielle. Ainsi, le styrène pourrait être obtenu par des procédés de catalyse homogène mettant en œuvre d’autres matières premières. On peut également envisager le remplacement à long terme du gaz naturel et du pétrole comme sources de matières premières de l’industrie chimique organique. L’utilisation d’un mélange d’hydrogène et de monoxyde de carbone (gaz de synthèse) apparaît comme une solution raisonnable: la production d’acide acétique par carbonylation du méthanol, lui-même obtenu à partir du gaz de synthèse, pourrait ainsi se substituer à l’oxydation de l’éthanal dont la synthèse s’effectue selon le procédé Wacker. Plusieurs grandes nations industrialisées ont déjà défini des programmes de recherche et de développement ambitieux sur la carbochimie et des applications sont déjà en cours de réalisation. Ainsi, dès 1983, une usine américaine a produit de l’anhydride acétique par une voie purement carbochimique qui met en œuvre un catalyseur hétérogène pour la synthèse du méthanol et un catalyseur homogène pour sa transformation en anhydride acétique.
Encyclopédie Universelle. 2012.